Aldosterone

Introduction
One important lipid found in our body is aldosterone, a hormone whose main function is to regulate salt in the body. Despite this, aldosterone is also involved in numerous processes, such as inflammation, fibrosis, renal injury, and heart failure.

Figure 1: A diagram illustrating the structure of aldosterone. Created using Chemsketch.
Aldosterone in Salt Regulation
The main function of aldosterone is the regulation of salts, such as sodium and potassium, in our body. When aldosterone is released, it binds to the mineralocorticoid receptor (MR), which can be found in the distal convoluted tubule, connecting tubule, and collecting duct of the kidney’s nephron.
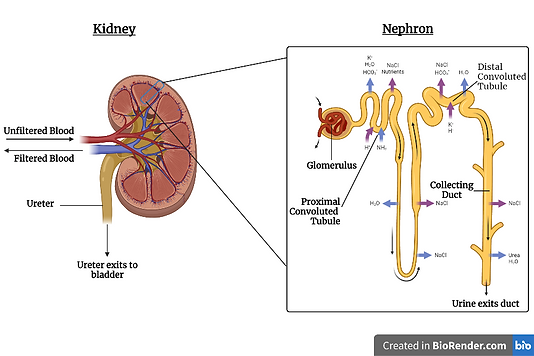
Figure 2: A diagram illustrating the structure of a nephron. The mineralocorticoid receptors (MRs) mentioned above can be found in the distal convoluted tubule, connecting tubule, and collecting duct. Created using BioRender.com.
The binding of aldosterone to an MR causes an increase in sodium reabsorption. This means that more salt will enter our body again, and less salt will be excreted via our urine. This binding of aldosterone to MR also results in the upregulation of a specific kinase known as the serum/glucocorticoid-regulated kinase (SGK1). This kinase phosphorylates a certain protein known as Nedd4-2. The phosphorylation of Nedd4-2 results in its inactivation. This, in turn, prevents it from ubiquitinating sodium chloride transporters located in the late distal convoluted tubule and the distal tubule. Ubiquitination is a biological process in which proteins are tagged with ubiquitin in order to be degraded. Since the transporters are not ubiquitinated, they will not be degraded and can successfully carry out sodium reabsorption (Brown, 2013).
Aldosterone can also cause the upregulation of potassium transporters, resulting in an increase in the removal of potassium in the kidney. One such transporter that is affected in this way is the renal outer medullary K+ channel (ROMK) (Zhang et al., 2022). Although normal aldosterone levels do not really contribute to the homeostasis of potassium levels in our body, large changes in either potassium or aldosterone levels will significantly impact the levels of the other. This is because high levels of potassium modulates the phosphorylation of MR, which thus ends up altering its activity (Penton et al., 2015). This is especially important to keep in mind when working with aldosterone therapeutics, as aldosterone levels will also affect how much potassium is reabsorbed and how much is removed from the body. If the potassium homeostasis is affected, this could lead to significant side effects, such as cardiac failure (El-Sherif & Turitto, 2011). In the kidneys, aldosterone can also contribute to chronic kidney disease.
Aldosterone in Chronic Kidney Disease
Chronic kidney disease (CKD) is a disease characterised by damaged kidneys, causing the malfunction of MR and thus the inability to filter blood properly. This results in the excess fluid and wastes from the blood accumulating into the body which, in turn, leads to other health problems such as heart disease and stroke. Therefore, such patients benefit from treatment using MR antagonists (MRAs). The renin-angiotensin-aldosterone system (RAAS) is activated in the early stages of CKD so as to maintain the glomerular filtration rate (GFR). This is important as it regulates blood volume and systemic vascular resistance. Chronic activation of the RAAS is maladaptive and gives rise to progressive renal injury. Angiotensin II, the major mediator of RAAS-induced renal injury, exerts glomerular effects and activates proinflammatory and profibrotic pathways. As a result, patients suffering from CKD are treated with either angiotensin-converting enzyme inhibitors (ACEIs), angiotensin type 1 receptor blockers (ARBs), or both. Aldosterone also damages organs, particularly the heart and kidneys, directly. Its pathophysiological actions are similar to those of angiotensin II (Spencer et al., 2020).
Elevated levels of aldosterone in plasma increase the risk of kidney injury. Treatment with MRAs is beneficial to patients suffering from renal disease as such antagonists evade any renal histopathological changes, and reduce proteinuria and blood pressure. Nonepithelial MRs predominantly give rise to the detrimental effects on the kidney that are caused by aldosterone. Despite this, such effects can arise irrespective of the effect of aldosterone on blood pressure (Spencer et al., 2020).
The harmful effects of MR activation are supported by a phenomenon known as “aldosterone breakthrough”. This occurs in patients receiving ACEI/ARB therapy whose concentration of aldosterone in plasma is initially reduced and later returns to or exceeds the concentration observed prior to such treatment. Such a phenomenon is linked to severe proteinuria as well as faster deterioration in renal function (Spencer et al., 2020).
Aldosterone in Inflammation
As previously mentioned, aldosterone is involved in inflammation. It stimulates this process via the following pathway, illustrated in Figure 3 below.
-
Aldosterone stimulates the enzymes glutathione oxidase and nicotinamide adenine dinucleotide phosphate [NADP(H)] oxidase in various cells.
-
These oxidases generate superoxide molecules (a type of reactive oxygen species).
-
Superoxide molecules activate the specific transcription factor known as nuclear factor kappa b (NF-κB).
-
Once NF-κB is activated, it moves into the nucleus and initiates the transcription of various genes that cause inflammation.
(Lawrence, 2009); (Brown, 2013)

Figure 3: A diagram illustrating how aldosterone can initiate inflammation through the activation of the transcription factor NF-κB. Created using BioRender.com.
The type of cell affects its response to inflammation. For instance, activated NF-κB in the heart activates numerous inflammatory pathways, resulting in various effects.
This is observed in sterile inflammation, a process triggered by cardiac damage. Such damage inhibits cardiac repair, resulting in an inflammatory response. This inhibition continues until the factor that triggers inflammation is no longer present. Once this point has been reached, inflammation no longer takes place and so, cardiac repair can be carried out. However, too strong of an inflammatory response may result in chronic inflammation.
This occurs when NF-κB transcription produces high levels of inflammatory cytokines. One such example is the tumour growth factor β (TGF-β). These inflammatory cytokines in turn recruit several immune cells, such as neutrophils and monocytes. These immune cells will then produce matrix metalloproteases (MMPs), which degrade the extracellular matrix of the heart. This extracellular matrix is what keeps the heart together, and this degradation can result in long-lasting cardiac injury. In addition, the degraded matrix promotes this inflammatory response, so this chronic inflammation is further enhanced by itself (Prabhu & Frangogiannis, 2016).
Inflammatory environments can also eventually lead to the generation of tumours. For example, the reactive oxygen species that are formed during aldosterone signalling (see Figure 3 above) can help further mutate the cancer cells via oxidative DNA damage. Another example is that some inflammatory cytokines that arise from NF-κB transcription can end up promoting cancer growth (Singh et al., 2019). Although the full details pertaining to the relationship between aldosterone and cancer are not quite known yet, it seems to be the case that aldosterone signalling induces mitochondrial dysfunction, which promotes the epithelial to mesenchymal transition (EMT). This EMT is one of the processes that lead to metastasis. Metastasis is the spread of cancer from its original body part to another part of the body, and it is this process that makes cancer malignant (Yuan et al., 2012; Brown, 2013).
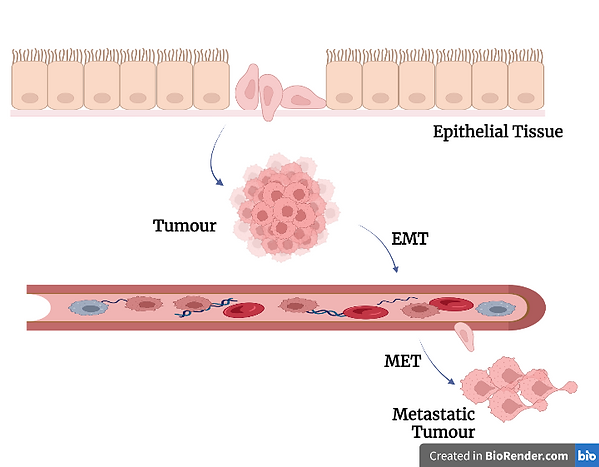
Figure 4: A diagram illustrating the process of epithelial to mesenchymal transition (EMT), briefly mentioned above. Created using BioRender.com.
Aldosterone in Fibrosis
Aldosterone is also involved in fibrosis, an essential pathological response to chronic inflammation. Fibrosis is the excessive deposition of proteins of the extracellular matrix (ECM) in the interstitium, a fluid-filled space between the organ and the surrounding tissue. This process takes place as a protective measure and is important for wound healing and tissue regeneration. However, there can be too much continuous pathological cardiac remodelling that occurs when heart tissue is damaged. This is usually coupled with the ECM deposition of fibrosis, and together they result in the distortion of the structure of the heart, impacting its function (Maruyama & Imanaka-Yoshida, 2022).
Fibrosis takes place when the production rate of collagen and the matrix exceeds their degradation rate via matrix metalloproteinases (MMPs). Moreover, aldosterone stimulates the expression of profibrotic molecules, which promote fibrosis. These include transforming growth factor-β1 (TGF-β1), plasminogen activator inhibitor 1 (PAI-1), endothelin 1 (ET-1), and placental growth factor (PGF) (Brown, 2013).
TGF-β1 promotes fibrosis by stimulating the cellular transformation of various cells to fibroblasts. It does so by increasing the synthesis of both matrix proteins and integrins, and decreasing the MMP production (Brown, 2013).
PAI-1 is a member of the serpin (serine protease inhibitor) superfamily. It inhibits the activation of plasminogen to plasmin by tissue plasminogen activator (tPA). This serpin prevents plasmin-mediated MMP activation and extracellular matrix degradation, thus promoting fibrosis and remodelling. Conversely, PAI-1 can exert anti-fibrotic effects. It does so either by retarding cellular infiltration and impending urokinase-type plasminogen activator (uPA) or by plasmin mediated activation and the release of latent growth factors. Aldosterone induces the expression of PAI-1 in the heart, however, a deficiency of this serpin in this organ enhances aldosterone-induced or angiotensin-II-induced fibrosis (Brown, 2013).
Aldosterone and salt stimulate an increase in the expression of ET-1. This, in turn, gives rise to cardiovascular fibrosis and hypertrophy. ET-1 increases collagen synthesis in cardiac fibroblasts through a TGF-β1-dependent mechanism. Furthermore, the expression of ET-1 in the heart and vasculature increases via mineralocorticoid and salt treatment. On the other hand, treatment with an endothelin-A-receptor blocker prevents both aldosterone-induced fibrosis and vascular fibrosis. Additionally, aldosterone modulates binding to a steroid-responsive element in the Edn1 gene, which encodes ET-1, in the medullary collecting duct cells of the kidney. Moreover, aldosterone stimulates the expression of PGF in atherosclerotic human vessels via an MR response element (Brown, 2013).
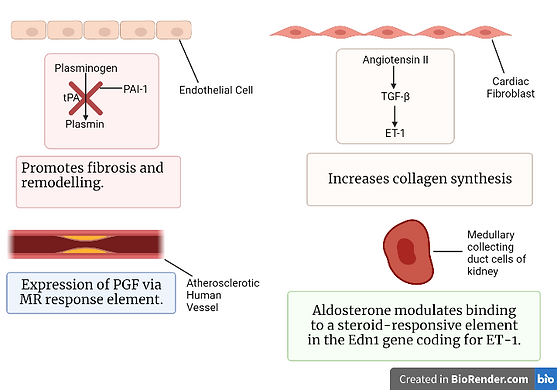
Figure 5: A schematic illustrating the process of fibrosis. Created using BioRender.com.
Aldosterone and the Cardiovascular System
Aldosterone is also linked to the cardiovascular system. Indeed, it has been found that more aldosterone is released when a person has congestive heart failure than when a person does not. Whilst aldosterone’s main function is the regulation of salt levels in the body, as described above, it has been shown that it can also damage, or worsen the damage of, the cardiovascular system. In fact, it has been found that people who have a higher than average amount of aldosterone in their body have a higher cardiovascular risk than those people who have high blood pressure. This is due to the fact that an increase in aldosterone levels can have multiple problematic effects on the heart. For example, increased levels of aldosterone can increase blood pressure, promote cardiac fibrosis, and cause left ventricular hypertrophy, which is when the main pumping chamber of the heart thickens, causing an increased pressure in the heart.
Now, onto how the body contributes to the high levels of aldosterone that reach the heart.
When the myocardium, which is the muscle of the heart, is damaged, more aldosterone is produced by the myocardium itself. The aldosterone produced can then bind to MR receptors, which are even found on the heart, and leads to cardiac fibrosis. When angiotensin II binds to the angiotensin type 1 (AT1) receptors, more aldosterone is secreted. Angiotensin II mediates the enzyme aldosterone synthase, which is the enzyme responsible for the synthesis of aldosterone. In turn, an increase in the aldosterone levels causes the upregulation of the translation of the gene that codes for the angiotensin-converting enzyme. This enzyme is responsible for the production of angiotensin II. Therefore, there is a positive feedback loop in this reaction, which results in constant increase in aldosterone, which leads to further cardiovascular damage (Nappi & Sieg, 2011).
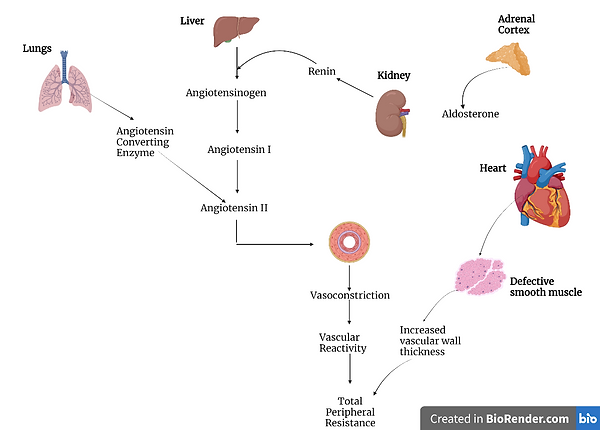
Figure 6: A diagram illustrating how aldosterone and angiotensin II can result in cardiovascular damage. Made using BioRender.com.
Conclusion

Figure 7: A diagram illustrating the different effects of aldosterone on our body. Created using BioRender.com.
The above highlights the different roles of aldosterone; its role in salt regulation, sodium and potassium ions in particular, the increased risk of kidney injury, its effects on the enzymes glutathione oxidase and NADP(H) oxidase, as well as its impact on the inflammatory response. Additionally, this hormone stimulates the expression of profibrotic molecules, promoting fibrosis. Aldosterone can also have detrimental effects on the cardiovascular system, including increased blood pressure, left ventricular hypertrophy, and cardiac fibrosis. This all comes to show the various effects of aldosterone and how crucial aldosterone homeostasis is in our body.
References:
Al‐Hashedi, E. M., Zhao, X., Mohammed, A. A., Juvenal, H., & Yu, J. (2022). Serum aldosterone effect on left ventricular structure and diastolic function in essential hypertension. The Journal of Clinical Hypertension, 25(2), 213–222. https://doi.org/10.1111/jch.14612
Brown, N. J. (2013). Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nature Reviews Nephrology, 9(8), 459–469. https://doi.org/10.1038/nrneph.2013.110
El-Sherif, N., & Turitto, G. (2011). Electrolyte disorders and arrhythmogenesis. Cardiology Journal, 18(3), 233–245. https://pubmed.ncbi.nlm.nih.gov/21660912/
Lawrence, T. (2009). The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harbor Perspectives in Biology, 1(6), a001651. https://doi.org/10.1101/cshperspect.a001651
Maruyama, K., & Imanaka-Yoshida, K. (2022). The pathogenesis of cardiac fibrosis: A review of recent progress. International Journal of Molecular Sciences, 23(5), 2617. https://doi.org/10.3390/ijms23052617
Nappi, J., & Sieg. (2011). Aldosterone and aldosterone receptor antagonists in patients with chronic heart failure. Vascular Health and Risk Management, 2011(7), 353–363. https://doi.org/10.2147/vhrm.s13779
Penton, D., Czogalla, J., & Loffing, J. (2015). Dietary potassium and the renal control of salt balance and blood pressure. Pflugers Archiv: European Journal of Physiology, 467(3), 513–530. https://doi.org/10.1007/s00424-014-1673-1
Prabhu, S. D., & Frangogiannis, N. G. (2016). The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circulation Research, 119(1), 91–112. https://doi.org/10.1161/circresaha.116.303577
Singh, N., Baby, D., Rajguru, J. P., Patil, P. B., Thakkannavar, S. S., & Pujari, V. B. (2019). Inflammation and cancer. Annals of African Medicine, 18(3), 121–126. https://doi.org/10.4103/aam.aam_56_18
Spencer, S., Wheeler‐Jones, C., & Elliott, J. (2020). Aldosterone and the mineralocorticoid receptor in renal injury: A potential therapeutic target in feline chronic kidney disease. Journal of Veterinary Pharmacology and Therapeutics, 43(3), 243–267. https://doi.org/10.1111/jvp.12848
Yuan, Y., Chen, Y., Zhang, P., Huang, S., Zhu, C., Ding, G., Liu, B., Yang, T., & Zhang, A. (2012). Mitochondrial dysfunction accounts for aldosterone-induced epithelial-to-mesenchymal transition of renal proximal tubular epithelial cells. Free Radical Biology and Medicine, 53(1), 30–43. https://doi.org/10.1016/j.freeradbiomed.2012.03.015
Zhang, D.-D., Zheng, J.-Y., Duan, X.-P., Lin, D.-H., & Wang, W.-H. (2022). ROMK channels are inhibited in the aldosterone-sensitive distal nephron of renal tubule Nedd4-2-deficient mice. American Journal of Physiology. Renal Physiology, 322(1), F55–F67. https://doi.org/10.1152/ajprenal.00306.2021