
HSP90
What are heat shock proteins?
Heat shock proteins were discovered by the Italian scientist Ferruccio Ritossa. By mistake, Ritossa had left flies (Drosophila melanogaster) at a higher temperature than usual (Hoter, El-Sabban, & Naim, 2018). He noticed puffing in specific regions of chromosomes which he reasoned that it had occurred due to an increase in gene expression (Hoter, El-Sabban, & Naim, 2018). These novel proteins were hence named heat shock proteins. According to Miller and Fort (2018), heat shock proteins (HSPs) are a large family of molecular chaperones that have roles in protein maturation, re-folding and protein degradation. Some HSPs are constitutively expressed whereas others are upregulated in response to stressful stimuli. HSPs are evolutionary conserved molecular chaperones with essential roles in cell survival and development. HSPs are categorized into two types. The first type are ATP-independent HSPs. These are chaperones of a molecular mass between 8 and 28 kDa. Examples of these types of HSPs include ubiquitin, a-crystallins and HSPB1. The second type of HSPs are ATP-dependent HSPs with a molecular mass between 40 and 105 kDa. Examples of these include HSP90 and HSP70.
What is their function and why are they important?
Jolly and Morimotot (2020) claim that the function of HSPs is to refold misfolded proteins thus, contributing to the maintenance of cellular homeostasis. HSPs are able to interact with various proteins to assist in their folding notably during cell stress to prevent the formation of misfolded and damaged proteins. HSPs aid in the recovery from stress by two mechanisms which are protein refolding and protein degradation. These two mechanisms are essential in restoring protein homeostasis and promoting cell survival.
According to Miller and Fort (2018), HSPs also provide thermotolerance in all organisms. Furthermore, HSPs provide protection from insults including hypoxia and cytotoxic exposure. Currently, HSPs are known to perform critical functions in both stressed and unstressed conditions. In normal conditions, HSPs promote proper embryonic and postnatal development of multiple organ systems especially the nervous system. During embryonic development, neuronal and glial progenitors must survive a hypoxic microenvironment whilst carrying out energy demanding processes such as cell migration and outgrowth of neurons. Since HSPs are induced by stress, they may have a role in neurodevelopment. In fact, studies have demonstrated that HSPs regulate pathways including the PI3K/AKT and RhoA which are involved in regulating cell growth and cell migration.
HSP90 aid in protein folding through the process known as the chaperone cycle which will be discussed in "HSP90 function and mode of action"
Heat Shock Protein 90: Structure + isoforms
HSP90 homologues are comprised of three conserved domains: N-terminal domain (NTD), C-terminal domain (CTD), and middle domain (MD) each with a specific function. There is a variable charger link domain present only in eukaryotes that links the NTD to the MD. HSPs are often classified based on their weight.
N-terminal Domain:
NTD contains an ATP binding motif so it is called the nucleotide-binding site. The ATP binding motif is highly conserved with the GHKL (Gyrase, HSP90, histidine kinase, and MutL) superfamily but differs from HSP70 or protein kianses. HSP90 ATPase activity is necessary for the chaperone cycle and for binding to client proteins. NTD has been utilized as a major target for HSP90 inhibitors such as Geldanamycin (GA) and Radicicol (RD) which compete with ATP for the binding pocket.
Charged Linker Region (CR):
Eukaryotic HSP90 contains a CR (absent in prokaryotes) which links the NTD to the MD. This domain has variable length and amino acid composition but is typically highly charged. This region is attributed to increased flexibility and dynamicity to help it manoeuvre in the crowded eukaryotic cell. Some studies suggest the charged region as a modulator of the chaperone activity, others neglect its potential influence on the HSP90 functions.
Middle Domain (MD):
MD modulates HSP90 function by binding the γ-phosphate of ATP specified for the NTD, to modulate its ATPase activity. Additionally, several studies demonstrated that this domain is implicated in binding co-chaperones like Aha1 and interacting with client proteins.
C-Terminal Domain (CTD):
CTD contains two key sites for calmodulin binding and another for HSP90 homodimersiation. CTD also contains a nucleotide-binding site which opens following the occupancy of the N-terminal site and serves as allosteric regulator of the N-terminal ATPase activity. The CTD domain differs from the NTD in terms of ligand specificity. The N-terminal binding site was found specific to adenosine nucleotides containing an intact adenine ring (such as nicotinamide adenine dinucleotides and adenosine polyphosphate alarmones), while the C-terminal binding site was much more unspecific by interacting with both purine and pyrimidine nucleotides (such as GTP and UTP nucleotides).
2′,3′-O-(2,4,6-trinitrophenyl)-nucleotides (TNP-ATP, TNP-GTP) and pyrophosphate were able to bind the C-terminal binding site with no need for occupied N-terminal site. Drugs like novobiocin (NB) and epigallocatechin-3-gallate (EGCG) could affect the function of HSP90 by targeting the CTD.
The CTD is included in the chaperone activity of HSP90 towards transcription factors and vesicular stomatitis virus G-protein. Targeting the CTD by the monoclonal anti-hsp90 antibody could hinder the interaction of HSP90 with many protein clients including steroid receptors and actin filaments.
Isoforms:
There are two main mammalian isoforms of HSP90, localized in the cytoplasm; the inducible form Hsp90α and the constitutive form HSP90β. Although these cytoplasmic HSP90 isoforms have recently shown distinctive functions, most reviews do not distinguish them due to their remarkable structural and functional similarity. Therefore, the name HSP90 has been used for both HSP90 α and β unless indicated. Studies dealing with the differentiation between cytoplasmic HSP90 isoforms, revealed that although both isoforms very often form dimers, HSP90α tends to dimerize frequently compared to HSP90β. In addition, the α and β isoforms vary in specific regions along the protein sequence, suggesting diverse functions of the two isoforms.
See Figure 1 for an image outlining the different roles of HSP90 isoforms.
Dimerization:
Dimerization is essential for proper functioning of HSP90. Mammalian HSP90 is a phosphorylated dimer containing 2–3 covalently-bound phosphate molecules per monomer. Structural studies revealed that dimeric forms of the protein have elongated structure. As a hydrophobic chaperone, heat shock increases its hydrophobicity. Another aspect is because HSP90 has an ATP binding site in its NTD, self-phosphorylation is likely to occur with profound conformational changes. Interestingly, although well known to have an ATP binding domain, HSP90 was found to have both ATPase and GTPase activity.
Stability:
Heat Shock Proteins are capable of withstanding higher temperatures than typical proteins and resist denaturation due to extreme temperatures and conditions. This resistance is attributed to their 3D structure that contains more abundant and stronger hydrophobic packing, hydrogen bonds, enhanced secondary proteins structure, and helix dipole stabilization.
The formation of fibrils often accompanies diseases, the most well-known of which include Alzheimer's disease, Type II diabetes, and spongiform encephalopathies (for example, Mad Cow Disease and Creutzfeldt-Jakob Disease/CJD).
Amyloid fibrils are formed when normally soluble proteins assemble and aggregate to form insoluble fibers which are resistant to degradation, via a process of polymerisation. These fibrils are deposited extracellularly in the tissues and result in pathogenic effects. Such agglomeration of fibrils is inherently stable, with structural studies revealing that these are mainly composed of β-sheet structures in a cross-β conformation. As such, the fibrils are self-assembled and proteins themselves can fold into alternative conformations which allows for this self-assembly.
There are 3 main proposed models for the self-assembly of amyloid fibrils from their monomers, these being:
The refolding model
The natively disordered model
The gain-of-interaction model
The refolding model is the most well-known model and it describes a situation wherein a protein refolds from its native state to an amyloid-forming state.
The natively disordered model suggests that the protein is natively disordered and folds directly into an amyloid-forming state.
The gain-of-interaction model is where part of one amyloid protein monomer becomes a domain in another amyloid protein monomer which eventually forms the amyloid fibril.
It should also be noted that while, on the whole, amyloid fibrils of the same protein form consistent structures, other fibril features such as morphology and properties can vary according to exposure to different environmental conditions during growth. Hence, minute changes to the growth conditions can have a significant effect on the potential cytotoxicity of the formed amyloid fibril.
The reasons why amyloid fibrils cause diseases are still unclear. In some cases, the deposits physically disrupt tissue architecture, suggesting disruption of function by some bulk process. An emerging consensus implicates prefibrillar intermediates, rather than mature amyloid fibers, in causing cell death, particularly in neurodegenerative diseases.
MECHANISMS OF TOXICITY:
Calcium dysregulation has been observed to occur early in cells exposed to protein oligomers. These small aggregates can form ion channels through lipid bilayer membranes and activate NMDA and AMPA receptors. Channel formation has been hypothesized to account for calcium dysregulation and mitochondrial dysfunction by allowing indiscriminate leakage of ions across cell membranes. Studies have also shown that amyloid deposition is associated with mitochondrial dysfunction and a resulting generation of reactive oxygen species (ROS), which can initiate a signaling pathway leading to apoptosis.
Beta‐amyloid (Aβ) accumulation is also commonly associated with Alzheimer's disease. Aβ toxicity results in an age‐related increase in cholinergic loss and microglial activation in the brain, along with neuronal loss. Studies have also shown that age plays a crucial role in susceptibility of the brain to certain Alzheimer's pathologies including accumulation of beta-amyloid.
All these mechanisms of toxicity are likely to play a role. In fact, the aggregation of a protein generates a variety of aggregates, all of which are likely to be toxic to some degree. The oligomers have also been reported to interact with a variety of molecular targets. The misfolded nature of protein aggregates causes a multitude of aberrant interactions with a multitude of cellular components, including membranes, protein receptors, soluble proteins, RNAs and small metabolites. Hence, it is unlikely that there is a unique mechanism of toxicity or a unique cascade of cellular events.
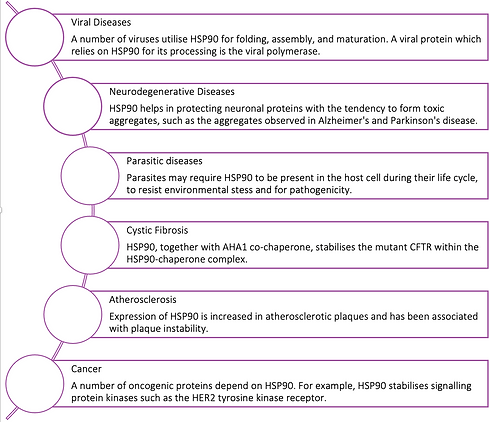
Figure 1
The Role of HSP90 in Disease
HSP90 function and mode of action
Functions:
HSP90 family has a multitude of different functions which can be attributed to the various isoforms. The functions of HSP90 are summarised below and are divided into sections pertaining to each isoform.
Cytosolic HSP90 – HSP90α/β
Chaperone Function – cytosolic HSP90 proteins have been found to assist protein folding and prevent protein aggregation. Specific examples include: stabilising heat-denatured proteins such as citrate synthase and protein kinase CK-II Preventing an increased aggregation of CK-II mild aggregates.Interacts with other chaperones to help denatured luciferase and β-galactosidase in refolding. Works with HSP40 and HSP70 and co-chaperones in refolding processes of denatured proteins such as steroid receptors and protein kinases (Hoter, El-Sabban, & Naim, 2018).
Cell Signalling – HSP90 is a central chaperone in cell signalling. It has been found in complex with many protein kinases such as the c-Src tyrosine kinase, of which it brings about hypo-phosphorylation and results in a lack of protein kinase activity. HSP90 has been found to help the Raf kinase to associate with the cell membrane, and to protect kinases from phosphatases and proteasomal degradation (Hoter, El-Sabban, & Naim, 2018).
Cytoskeleton – cytosolic HSP90 has been described as a molecular glue because of its adhesion properties and large quantity in the cytoplasm. HSP90 plays a big role in the organisation and maintenance of the cytoskeleton. Specifically, HSP90 has been found to modulate actin filament bundling, and interact with tubulin and protect it from heat denaturation. Moreover it was reported to interact with various other cytoskeletal structures including microtubules, and to protect myosin from heat stress (Hoter, El-Sabban, & Naim, 2018).
Nuclear Functions – HSP90 nuclear localisation can be up to 10% of the total cytosolic HSP90 and increases when the cell is under heat stress. HSP90 associates with DNA, RNA and histones to modulate DNA structure and plays a role in RNA synthesis and processing. HSP90 also interacts with many transcription factors, forming complexes with zinc-finger proteins, helix-loop-helix proteins, hypoxia-inducible factor 1α and heat-shock factor 1 (Hoter, El-Sabban, & Naim, 2018).
Cell cycle and cellular differentiation – HSP90 interacts with co-chaperones to fold several cell cycle-related protein kinases such as the cyclin-dependent kinase CDK4. The expression profile of HSP90 isoforms differs according to the stage of the cell cycle (Hoter, El-Sabban, & Naim, 2018).
GRP94
Protein Folding – the main role of GRP94 is to encourage the folding and assembly of secretory and membrane proteins (Hoter, El-Sabban, & Naim, 2018).
Calcium homeostasis – GRP94 is a calcium binding protein that interacts with other ER chaperones to mediate calcium homeostasis in the ER and other areas in the cell (Hoter, El-Sabban, & Naim, 2018).
Er quality control – In the ER, a network of chaperones work together to produce correctly folded secretory and membrane proteins. The GRP94 protein plays several roles in this network including interacting with other proteins in the ER protein folding machinery, chaperoning protein folding intermediates, working to target misfolded proteins, and in calcium storage (Hoter, El-Sabban, & Naim, 2018).
ER stress – GRP94 is involved in the unfolded protein response (UPR). Its actions help to increase the ER folding capacity, suppress apoptosis, and lessen metabolic stress when the cell is under stress (Hoter, El-Sabban, & Naim, 2018).
TRAP1
Its role in mitochondria – TRAP1 plays a big role in maintenance of mitochondrial integrity and protection against mitochondrial apoptosis. Moreover, it reduces reactive oxygen species (ROS) levels in mitochondria and protects against cell death induced by high ROS levels. Moreover, TRAP1 protects against protein aggregation inside the mitochondria, and aids in protein folding (Hoter, El-Sabban, & Naim, 2018).
Protection against oxidative stress – TRAP1 protects against oxidative stress by interacting with several proteins involved in pore formation in the inner mitochondrial membrane. This is called a permeability transition pore, and an increased ROS under stress conditions, will cause these pores to open up and lead to a disturbed membrane potential (Hoter, El-Sabban, & Naim, 2018).
Extra-mitochondrial roles – TRAP1 molecules have been observed trans-located outside the mitochondria where they bind to extra-mitochondrial proteins such as the retinoblastoma protein, TNF receptor, and tumour suppressor EXT proteins. During mitosis, TRAP1 interacts with the retinoblastoma protein to help its folding (Hoter, El-Sabban, & Naim, 2018).
Mode of Action
ATPase activity
The principle of HSP90 mechanism of action is its ATPase activity whereby it undergoes a cycle between closed and open states. HSP90 is a flexible homodimer, and each monomer is composed of 3 structural domains: the NTD, MD and the CTD. When ATP binds to the ATP binding cleft of the NTD this sparks off a series of conformational changes. One such conformational change takes place when the ATP-lid of the NTD (a small part of it) trans-locates over the ATP binding pocket and then attaches to the NTD of the other monomer in the homo-dimer which results in a twisted and compacted homodimer. This process brings the N- and M- domains closer to allow for final formation of the ‘split ATPase’ site. After ATP hydrolysis the N domains of the HSP90 homodimers dissociate and release ADP and Pi and the HSP90 returns to its original open conformation (Hoter, El-Sabban, & Naim, 2018).
Chaperone cycle
The HSP90 family members express their folding function of client proteins through a complex process known as the HSP90 chaperone cycle. A fully comprehensive understanding of this chaperone cycle is still not known. The HSP90 chaperone machinery requires cooperation of different molecules such as co-chaperons, partner proteins and immunophilins that concertedly act to aid in efficient protein folding by HSP90 proteins. A summary of the mechanism of protein folding by HSP90 is as follows. First, the client protein in its mid-folded or nascent polypeptide state is complexed with HSP70/HSP40/ADP which protects it from aggregation. This protein complex is further stabilised by HSP70-interacting protein or Bcl2 which enables the change from ADP to ATP. HSP90 then binds the client protein that is bound with the HSP70/HSP40 protein complex to begin its facilitative actions. An adaptor protein known as HSP90-HSP70 organising protein helps the interaction between HSP90 and HSP70. Additional co-chaperons such as p50 or Cdc37 then help loading client kinases onto HSP90. During client protein loading, co-chaperons such as immunophilins FKBP51/52 are added to the homodimer as an activated heteroprotein complex is brought together and the HSP70, the HSP70-interacting protein, and the HSP90-HSP70 organising protein are all released from the complex. Binding of ATP to the NTD of HSP90 in the heteroprotein complex then switches HSP90 from the ‘open state’ to the ‘closed state’. At this stage, if an inhibitor is present it can act to compete with ATP for the NTD binding site and results in protein degradation by the proteasome. If there are no inhibitors present to interfere with this folding cycle, other co-chaperones such as Aha1, may associate induces ATP hydrolysis and supporting proper folding of the bound client and enable release of other co-chaperons and immunophilins (Hoter, El-Sabban, & Naim, 2018).
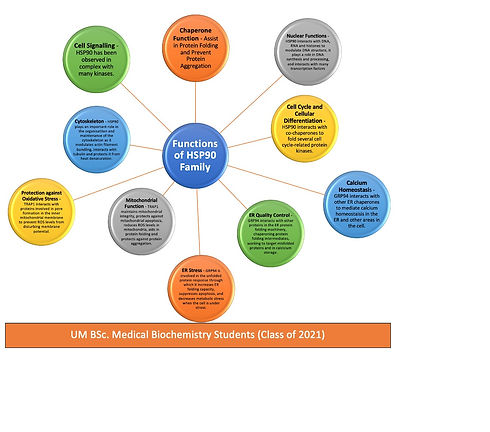
Figure 2
Summary of the Functions of the HSP90 Isoforms.
Role in Disease and other Health Implications
The Role of HSP90 in Cancer
Cancer is a complex disease characterised by uncontrolled proliferation, self-sufficiency in growth, limitless replication and tissue invasion (Miyata et al., 2013). Many of these processes are a result of the activation of oncogenic proteins that depend on the molecular chaperone HSP90 (Miyata et al., 2013). For instance, Hsp90 is required for the stability of signalling protein kinases such as the HER2 tyrosine kinase receptor that are required for efficient cell proliferation (Miyata et al., 2013). The HSP90 chaperone machinery also protects many oncogenic proteins like Raf and CDK4 from degradation and misfolding (Miyata et al., 2013). In fact, high expression of HSP90 has been detected in almost all types of cancer (Chatterjee & Burns, 2017).
For this reason, many HSP90 inhibitors have been used as chemotherapeutical agents to supress the functions of many oncogenic proteins. Natural products like geldanamycin (GA) and the antibiotic radicicol (RA) isolated from Streptomyces hygroscopicus and Monosporium bonorden respectively, have both exhibited significant suppression in tumour growth (Chatterjee & Burns, 2017). Since these have a similar structure to adenosine triphosphate (ATP), they are able to bind and block the ATP binding site of HSP90, rendering the chaperone inactive (Chatterjee & Burns, 2017). This then leads to the depletion of its oncogenic clients promoting cell death.
The Role of HSP90 in Neurodegenerative diseases
HSP90 helps in protecting neuronal proteins with the tendency from toxic aggregates. HSP90 works against Hprotein aggregation seen in neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease.
It is believed that in Alzheimer’s disease HSP90 promotes the hyperphosphorylation of tau protein (tau-p) through the activation of kinases leading to the formation of neurofibrillary tangles. When targeted by inhibitors, HSP90 lowered p-tau levels through a mechanism facilitated by tau ubiquitin ligase; carboxy terminus of Hsp70–interacting protein (CHIP).
Preliminary in vitro experiments for Parkinson’s disease revealed that HSP90 (and even HSP70/40) could suppress Aβ amyloid self-assembly. This suggests that the induction of chaperones’ overexpression by pharmacological substances is a prospective therapeutic pathway for neurodegenerative diseases.
Role of HSP90 in infectious diseases:
Viral Diseases:
Viral protein homeostasis is an essential process that occurs in viruses in order to sustain them and allow for their proliferation. Studies done in this area are suggesting that HSP90 appears to be involved in this process through its role in various stages of the viral lifecycle. In fact, a sizeable number of viruses have been observed to utilise HSP90 for folding, assembly, and maturation. Of the proteins which viruses possess, a protein that relies and requires HSP90 for its processing is the viral polymerase, a protein essential for viral replication. An example of this is the Reverse Transcriptase of duck Hepatitis B virus (DHBV) which requires HSP90 in conjunction with other chaperones (such as HSP70 and HSP40) and co-chaperones (e.g. HOP and p23) to act as substrate release factors and to support the incorporation of the pre-genome RNA (pgRNA) into nucleocapsids. Apart from DHBV, Influenza virus A also requires HSP90 for replicating its genome. It is suggested that the need for HSP90 by viruses for infection is a result of the high translation rates of viral proteins in addition to high mutation incidence and high conformational and proteolytic events. Hence, such stressful events and processes require HSP90 to act as a buffering system to ensure the integrity and stability of the whole system.
2. Parasitic diseases:
It has been found that Plasmodium falciparum, a malarial parasite, needs HSP90 in the human host in order to be able to resist the environmental stress such as pH and elevated temperature fluctuations in the host. Similarly, Leishmania donovani, responsible for Leshmaniasis, requires HSP90 during its lifecycle and for pathogenicity. In fact, studies found that as much as 3% of the total protein content of the promastigote (protozoan extracellular form) stage is composed of HSP90. In addition, there are around 17 homologues for the HSP90 gene in Leishmania major which are thought to be involved in pathogenesis. In Pristionchus pacificus it has been reported that HSP90 seems to affect its morphological features.
3. Role of HSP90 in other diseases:
HSP90 has been implicated in the pathogenesis of other serious diseases such as cystic fibrosis, atherosclerosis, and diabetes.
In cystic fibrosis, HSP90 was found to interact with the wild-type cystic fibrosis receptor (CFTR) protein as well as the mutated form. As part of the HSP90-protein complex, the AHA1 co-chaperone appears to play a key function in stabilising the mutant CFTR within the HSP90-chaperone complex. Knockdown of AHA1 has been found to lead to a high number of mutant CFTR channels being transferred to the plasma membrane which suggests that the AHA1 co-chaperone mediates processing and degradation of the mutant CFTR by blocking its escape from the chaperone cycle.
In patients with atherosclerosis, expression of HSP90 is increased in atherosclerotic plaques and has been associated with plaque instability. Interestingly, HSP90-inhibiting drugs could diminish plaque formation by suppressing the migration and proliferation of vascular smooth muscle cells. In other studies it was reported that the high HSP90 expression is not restricted to the atherosclerotic plaques. In fact, the serum of atherosclerotic patients contained elevated HSP90 levels presumably inducing immune response. Hence, HSP90 has been considered as a possible target auto-antigen in the pathogenesis of carotid atherosclerosis.
References
Bohush, A., Bieganowski, P., & Filipek, A. (2019). Hsp90 and its co-chaperones in neurodegenerative diseases. International journal of molecular sciences, 20(20), 4976.
Chatterjee, S., & Burns, T. F. (2017). Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. International journal of molecular sciences, 18(9), 1978. https://doi.org/10.3390/ijms18091978
Dickey, C. A., Kamal, A., Lundgren, K., Klosak, N., Bailey, R. M., Dunmore, J., Ash, P., Shoraka, S., Zlatkovic, J., Eckman, C. B., Patterson, C., Dickson, D. W., Nahman, N. S., Jr, Hutton, M., Burrows, F., & Petrucelli, L. (2007). The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. The Journal of clinical investigation, 117(3), 648–658. https://doi.org/10.1172/JCI29715
Evans, C. G., Wisén, S., & Gestwicki, J. E. (2006). Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1-42) aggregation in vitro. The Journal of biological chemistry, 281(44), 33182–33191. https://doi.org/10.1074/jbc.M606192200
Hoter, A., El-Sabban, M. E., & Naim, H. Y. (2018). The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. International Journal of Molecular Sciences, 19(9). https://doi.org/10.3390/ijms19092560
JollY, C., & Morimoto, R. I. (2000). Role of the heat shock response and molecular chaperones in oncogenesis and cell death. JNCI : Journal of the National Cancer Institute, 92(19), 1564-1572. doi:10.1093/jnci/92.19.1564
Miller, D. J., & Fort, P. E. (2018). Heat shock proteins regulatory role in neurodevelopment. Frontiers in Neuroscience, 12, 821. doi:10.3389/fnins.2018.00821
Miyata, Y., Nakamoto, H., & Neckers, L. (2013). The therapeutic target Hsp90 and cancer hallmarks. Current Pharmaceutical Design, 19(3), 347-365. doi:10.2174/138161213804143725
Roy, N., Nageshan, R. K., Ranade, S., & Tatu, U. (2012). Heat shock protein 90 from neglected protozoan parasites. Biochimica et biophysica acta, 1823(3), 707–711. https://doi.org/10.1016/j.bbamcr.2011.12.003
Schopf, F. H., Biebl, M. M., & Buchner, J. (2017). The HSP90 chaperone machinery. Nature reviews. Molecular cell biology, 18(6), 345–360. https://doi.org/10.1038/nrm.2017.20
Vartholomaiou, E., Echeverría, P. C., & Picard, D. (2016). Unusual Suspects in the Twilight Zone Between the Hsp90 Interactome and Carcinogenesis. Advances in cancer research, 129, 1–30. https://doi.org/10.1016/bs.acr.2015.08.001